
All of our experiments are carried out using three different molecular beam machines located on the second floor of ST Olin Chemistry Research Building, located on the central Cornell campus.
Endstation 1:
Our DOE-supported studies of the reaction dynamics of free radicals are carried out using Endstation 1.
This apparatus was originally constructed at Lawrence Berkeley Laboratory’s Advanced Light Source, and consists of two molecular beam sources crossing at right angles that can be rotated relative to a fixed mass spectrometer detector. In its original implementation, the apparatus used synchrotron radiation to ionize the neutral products from photodissociation and bimolecular reactions. Since this machine was moved to Cornell, we have been using pulsed VUV and XUV light produced by nonlinear conversion using high energy pulsed nanosecond lasers for photoionization.
Our new windowless VUV/XUV light source now installed on ES1 is illustrated schematically in the figure below (click on figure to expand). In our previous configuration, the VUV was dispersed from the much more intense residual UV and visible laser beams by passing the output through a MgF2 lens held off of the optic axis. The primary limitation was that transmission at λ < 120 nm is very inefficient for MgF2. While LiF is slightly better (but more easily damaged), transmission still falls to zero for λ ≤ 104 nm. Our new setup employs the two-photon Hg 6s → 7s resonance at 312.8 nm, with the VUV/XUV radiation (s-polarization) reflected efficiently at grazing angle. An early difficulty was experienced due to optical damage resulting from the high UV laser intensities, recent improvements to our vacuum system 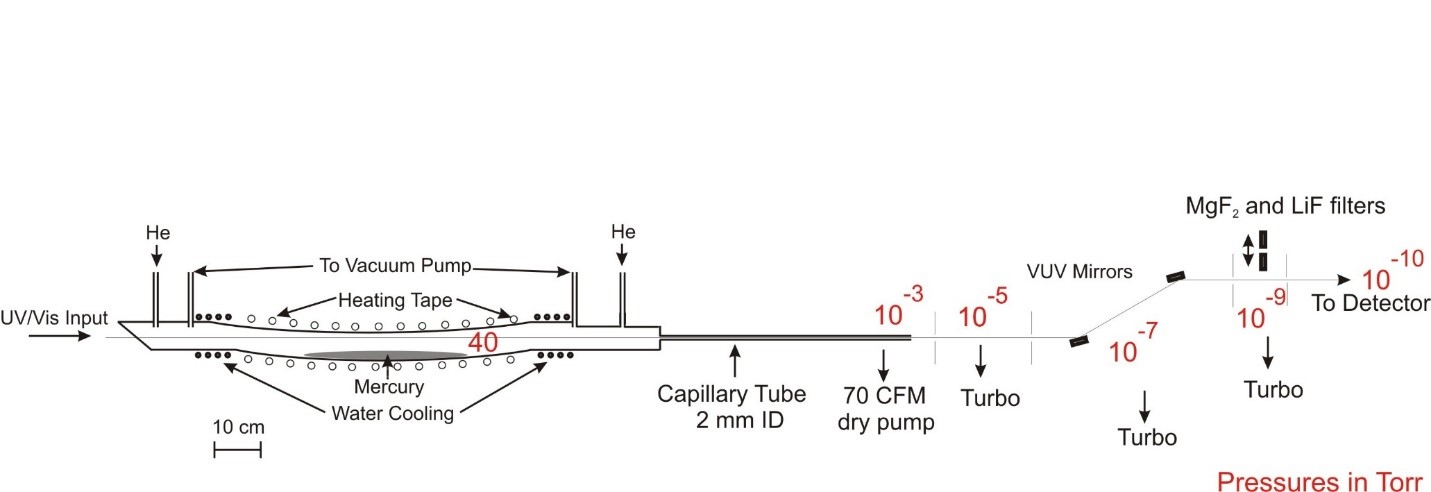 has made it possible to use high pulse energies (15 mJ/pulse at 312.8 nm) without any optical damage after many days of operation. The reflectivity of s-polarized light in the 100-120 nm range is ~ 60% for 75 degree incidence angle, whereas that near 312.8 nm (s-polarization) is < 0.2%. Therefore using two mirrors, the contrast ratio is approximately (60/0.2)2 > 90,000. A photo of the windowless beamline now in operation at Cornell on ES1 is shown below.
has made it possible to use high pulse energies (15 mJ/pulse at 312.8 nm) without any optical damage after many days of operation. The reflectivity of s-polarized light in the 100-120 nm range is ~ 60% for 75 degree incidence angle, whereas that near 312.8 nm (s-polarization) is < 0.2%. Therefore using two mirrors, the contrast ratio is approximately (60/0.2)2 > 90,000. A photo of the windowless beamline now in operation at Cornell on ES1 is shown below.

The use of unfocussed input lasers over long interaction lengths in Hg is most general and efficient if three independent input laser beams are each tuned near Hg resonances at the precise wavelengths where index matching may be achieved. In experiments involving O atom 3PJ→3S1 detection near 130 nm, the input wavelengths were 255 nm, 404 nm, and 777 nm. Thus the 255 nm laser is near the 3P1 resonance, the 255nm + 404nm corresponds to the 1So two photon resonance, and the 777 nm laser is only slightly detuned from an atomic resonance. Using such near-triple resonant methods, continuous wave (CW) Lyman-α has been produced. A simpler approach providing nearly the same intensities but with somewhat less flexible wavelength tuning can be achieved using two lasers, with one tuned to the 2-photon 6s→7s resonance near 312.8 nm. and a second near an atomic resonance above the ionization limit. A typical scheme used in our laboratory for production of light near 112 nm (11.0 eV) is shown in the figure below.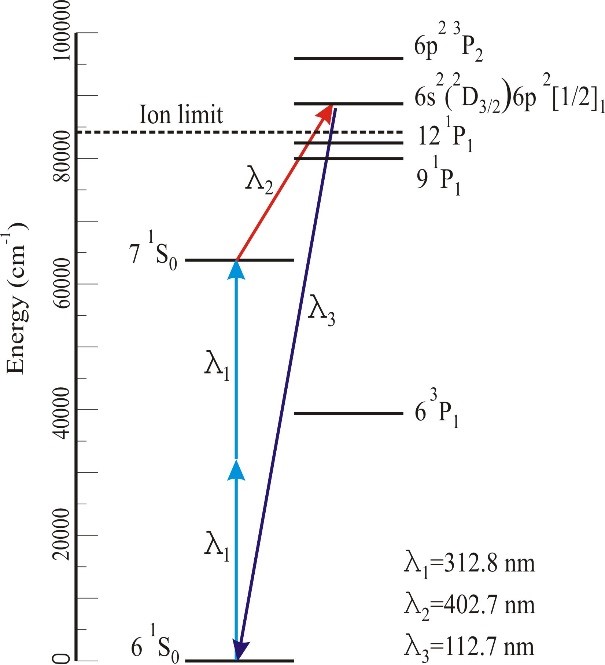
Prior to our previous research, it was not obvious that commercial multimode dye lasers could be used in an unfocussed configuration to produce VUV intensities comparable to those of Muller, who reported mJ pulse energies in the 130 nm range. However, as we have discussed in detail elsewhere, this approach works extremely well, facilitating “soft universal” photoionization detection of a range of interesting free radical species at 9.9 eV using the Cornell fixed detector, rotatable source crossed beams apparatus, originally used for NSF-supported studies of transition metal chemistry. The primary limitation of our original implementation of this technique was that it was not amenable to photoionization detection of the large number of key product species with ionization energies above 9.9 eV. Also, the relatively long neutral flight distance (25 cm) in the Cornell rotatable source apparatus led to small signal levels.
We have devoted great effort recenly to the development of a new windowless VUV beamline on ES1. In particular, the availability of intense pulsed light at 11.0 and 11.9 eV facilitates direct photoionization detection of HO2 (I.E. = 11.5 eV), as well as the products from bimolecular reactions of C2H5 with O2, namely C2H4 (I.E. = 10.5 eV), c- CH2CH2O (I.E. = 10.6 eV), and CH3CHO (I.E. = 10.2 eV). It also opens up direct photoionization detection of CH2O (I.E. = 10.9 eV), as well as ethane (I.E. = 11.5 eV), propane (10.9 eV), etc. Note that none of these products were detectable using 9.9 eV photons produced using our earlier high intensity light source.
Photoionization efficiencies typically increase upon tuning above the I.E., whereas the threshold for dissociative ionization typically lies 1-2 eV above the I.E. Therefore, photoionization detection of a wide range of products requires broad tunability of high- intensity pulsed light over the 9 eV to 12 eV range. It is important to note that it is not necessary in our experiments to carry out continuous photoionization yield scans— the wavelength only needs to be “line tunable” at discrete photon energies. After considerable experimentation, we have found what we believe to be an optimum configuration. First, to minimize complexity, two tunable dye lasers are employed, rather than three as used in some of our previous work, with one at the two photon resonance near 312.8 nm. The polarization of the VUV light is always the same as that of ν3 in four wave mixing schemes in Hg employing ν1 + ν2 = 63,928 cm-1. Following this scheme, a number of wavelengths may be readily selected, all with λ1 = λ2 = 312.8 nm, as follows: 9.5 eV (λ3 = 770 nm; LDS765 dye); 9.9 eV λ3 = 630 nm; DCM dye); 11.0 eV (λ3 = 404 nm; DCM dye + 1064 nm); and 11.9 eV (λ3 = 312.8 nm = λ1 =λ2). For 9.5-11.0 eV, we use a 532 nm-pumped Scanmate 2 dye laser and solid prisms to propagate λ3, and two separate sets of dye cuvettes and circulators for DCM and LDS765 dyes. The cuvettes may be readily switched (< 2 mins) between dye fundamentals (for 770 and 630 nm) as well as mixing with 1064 nm (for 404 nm) using removable keyed optical elements. Thus no optical realignment is required upon wavelength changes. The 11.9 eV beam is simply the third harmonic of the 312.8 nm laser and is obtained with the λ3 dye laser blocked. From our measurements, the 11.9 eV intensities using 13 mJ/pulse at 312.8 nm are within a factor of three of the most efficient mixing scheme at 9.9 eV, corresponding to >50μJ/pulse when a 1m lens is employed to reduce the diameter of the input laser to 1 mm in the Hg cell. Since the detector ionization region is 2 m away from this lens, the 11.9 eV laser spot size is essentially the same as that using collimated input laser beams. MgF2 and LiF windows are mounted on a vacuum manipulator and can be translated into the beam following the dichroic mirrors to suppress the 11.9 eV third harmonic for operation at 9.5, 9.9, and 11.0 eV.
Cornell Rotatable Source Apparatus:
This apparatus, designed and built at Cornell by the Davis group, is currently being used for NSF-supported studies of halogen oxide photochemistry and bimolecular reaction dynamics experiments. The photo on the left shows the source assembly just prior to insertion into the apparatus, taken around 1997. On the right is an old photo showing the differentially-pumped rotating ring being inserted into the apparatus (sitting on its side).  Previously, we used this apparatus for studies of transition metal
Previously, we used this apparatus for studies of transition metal  chemistry, such as reactions of neutral transition metal atoms with hydrocarbons such as methane. A representative publication can be viewed by clicking on the following link: 49-134nk3z
chemistry, such as reactions of neutral transition metal atoms with hydrocarbons such as methane. A representative publication can be viewed by clicking on the following link: 49-134nk3z
This apparatus shares some design elements with endstation 1 (above). The primary differences are that the source chambers in the Cornell machine are much larger, making it possible to install complex sources including laser ablation sources for transition metal chemistry experiments, as well as merged nozzle assemblies for production of polyatomic free radicals using reactants that would normally react thermally. For example, we have produced the OIO free radical, relevant to atmospheric chemistry, by the self reaction of OI produced by photolysis of O3 followed by reaction with CF3I, as described in detail below.
A photo of this apparatus in its current configuration for studies of halogen oxide chemistry is shown here.  Recently, we studied the photodissociation dynamics of OIO radicals induced by absorption of visible light. The OIO molecular beam was produced through the reaction OI + OI → OIO + I in a quartz tube reactor. The OI was produced by photolysis of O3 mixed with CF3I at 266 nm using the fourth-harmonic output of a Nd:YAG laser (Continuum 8020). The CF3I/O3 mixture was photolyzed within the quartz tube approximately 4 cm upstream of its exit. Upon photolysis, O3 dissociates predominantly to form O(1D2), which is subsequently quenched to O(3P) and reacts with an excess of CF3I to produce IO. The IO self-reaction occurs as the gas mixture travels through the remainder of the tube, yielding OIO. Following supersonic expansion from the end of the quartz tube, the molecular beam passes through a differentially pumped region and was collimated by two skimmers. The photodissociation laser was either the fundamental output from a Nd:YAG pumped Lambda-Physik Scanmate 2 dye laser (0.1 cm-1 bandwidth), or the second harmonic 532.1 nm of a Nd:YAG laser (1 cm-1 bandwidth).
Recently, we studied the photodissociation dynamics of OIO radicals induced by absorption of visible light. The OIO molecular beam was produced through the reaction OI + OI → OIO + I in a quartz tube reactor. The OI was produced by photolysis of O3 mixed with CF3I at 266 nm using the fourth-harmonic output of a Nd:YAG laser (Continuum 8020). The CF3I/O3 mixture was photolyzed within the quartz tube approximately 4 cm upstream of its exit. Upon photolysis, O3 dissociates predominantly to form O(1D2), which is subsequently quenched to O(3P) and reacts with an excess of CF3I to produce IO. The IO self-reaction occurs as the gas mixture travels through the remainder of the tube, yielding OIO. Following supersonic expansion from the end of the quartz tube, the molecular beam passes through a differentially pumped region and was collimated by two skimmers. The photodissociation laser was either the fundamental output from a Nd:YAG pumped Lambda-Physik Scanmate 2 dye laser (0.1 cm-1 bandwidth), or the second harmonic 532.1 nm of a Nd:YAG laser (1 cm-1 bandwidth).

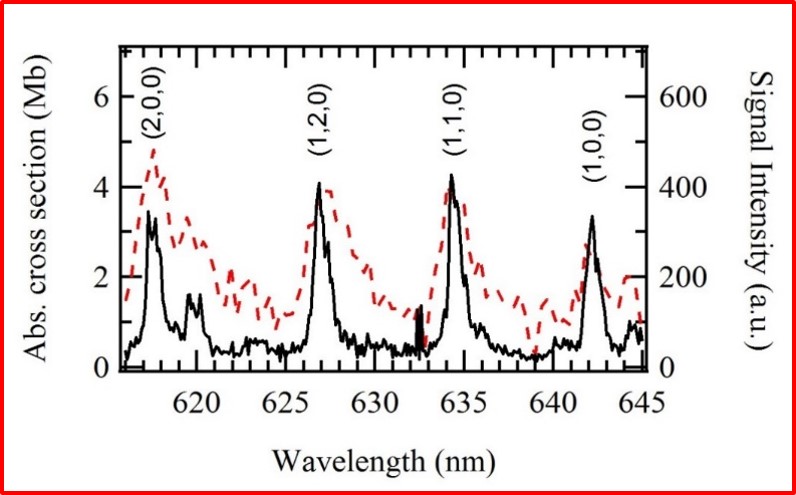
We found that OIO radicals dissociate exclusively to produce I + O2, with the O2 molecules produced in both their ground triplet states, as well as in the first excited singlet state. In the photo below, the I atom signal as a function of OIO excitation wavelength, recorded at a laboratory angle of 10 degrees relative to the OIO beam, is shown as a solid black line. The signal “action spectrum” for production of I atoms follows the known OIO absorption spectrum, indicated as a broken red line. Note that OIO molecules in a molecular beam are rotationally cold, so the lines are narrower than in a room temperature sample. Since OIO florescence is absent in this wavelength range, the quantum yield for production of I + O2 in this wavelength range is unity. We can also set the wavelength at a fixed excitation wavelength and measure the velocity distributions of the I atoms. This allows us to determine the kinetic energy distributions for the dissociation producing I + O2. From conservation of energy and linear momentum, by analyzing the arrival time distributions we can gain insight into the disposal of excess energy into the internal degrees of freedom of the product molecules, in this case O2. As can be seen in the translational energy distribution P(E), a broad range of O2 vibrational levels are populated in both the ground triplet and excited singlet states. 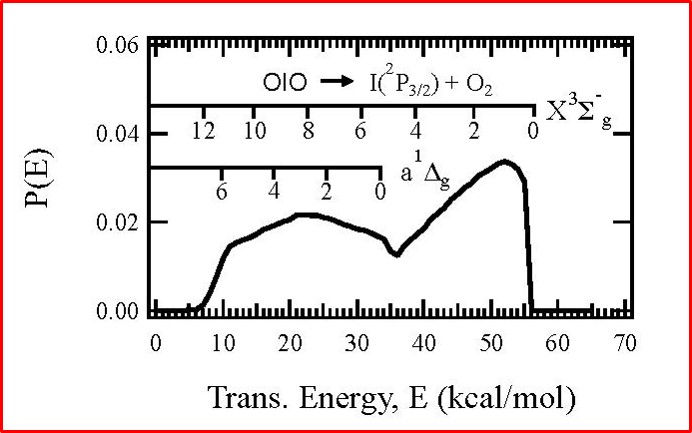
{kind=link}
Single Beam Apparatus:
We also have a single beam apparatus, pumped exclusively by turbomolecular pumps, that can be used for a variety of purposes. These include photodissociation experiments using a linear TOF mass spectrometer, and developing sources of free radicals for use on the other machines. This apparatus has a number of ports facilitating introduction of visible, UV, or VUV light. Recently we have been developing a new “coaxial VUV detection method” using this apparatus. A photo of the apparatus in its present configuration is shown below.